SISUKORD
GENEETIKA EHK DENT‘I HAIGUSE PÄRILIKKUS
- Madala molekulmassiga proteinuuria (LMW proteinuuria)
- Hüperkaltsuuria - kaltsiumi suurenenud eritumine uriiniga
- Neerukaltsinoos (nefrokaltsinoos) või neerukivid (nefrolitiaas)
- Hematuuria (tavaliselt mikrohematuuria)
- Fanconi sündroom ja madal seerumi fosfaatide tase
- Krooniline neeruhaigus ja neerupuudulikkus
- Muud sümptomid

Dent’i haigus on haruldane kaasasündinud neeruhaigus, mis põhjustab neerukivide teket ja sageli ka neerupuudulikkust. Sümptomid ilmnevad tavaliselt lapsepõlves, kuid need võivad jääda ka märkamatuks kuni täiskasvanueani.
Haiguse peamiseks tunnuseks on väikeste valgumolekulide esinemine uriinis koos hüperkaltsuuriaga (kaltsiumi suurenenud eritumisega uriini), kaltsiumikivide moodustumisega neerudes (nefrokaltsinoosi) ja korduvate neerukiviepisoodidega (nefrolitiaasiga).
Progresseeruvad neeruprobleemid viivad sageli neerupuudulikkuseni täiskasvanueas. Harvemini esinevad rahhiit st kaltsium-fosfaadihäiretest tingitud luumuutused ja kasvuhäired.
Haigus esineb tavaliselt ainult meestel, kuna selle pärilikkus on seotud sugukromosoomiga (vt allpool). Haiguse esinemissagedus võib olla alahinnatud, tõenäoliselt paljude patsientide kaebuste puudumise ja vähetuntuse tõttu. Haigust diagnoositakse sageli liiga hilja, kui neerufunktsioon on juba häirunud.
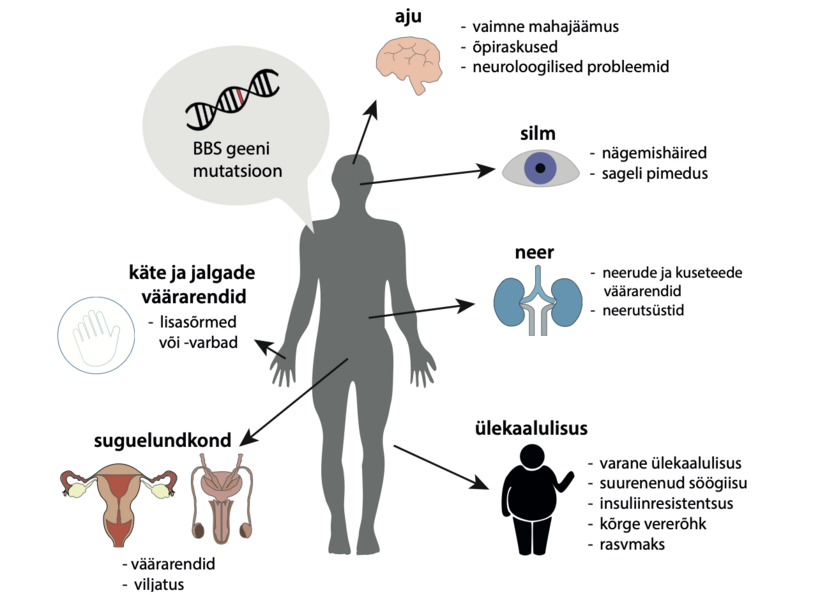
Dent’i haigus on kaasasündinud, st geneetiliselt pärandatud, monogeenne haigus, mis tähendab, et selle esinemine on tingitud ühe konkreetse geeni (antud juhul kas CLCN5 või OCRL1 geeni) mutatsioonist ja kuulub haiguste rühma, mida nimetatakse tubulopaatiateks. Tubulopaatiad on haigused, mille puhul defekt mõjutab neerutorukesi(vt allpool).
Eristatakse proksimaalseid ja distaalseid või segatubulopaatiaid, sõltuvalt sellest, millist tuubuluse osa on kahjustatud. Dent’i haiguse puhul mõjutab toime defekt peamiselt proksimaalseid neerutorukesi. Nendes neerutorukese lõikudes imenduvad paljud ained, mis on glomeerulites (neerufi ltrites) fi ltreeritud, uuesti, sest organism vajab neid endiselt. Geenid CLCN5 ja OCRL1 kodeerivad spetsiifi lisi ensüüme, mis on vajalikud madalmolekulaarsete valkude ja mineraalainete, nagu kaalium, fosfaat, kaltsium ja bikarbonaat, tagasiimendumiseks.
Kas te teadsite seda?
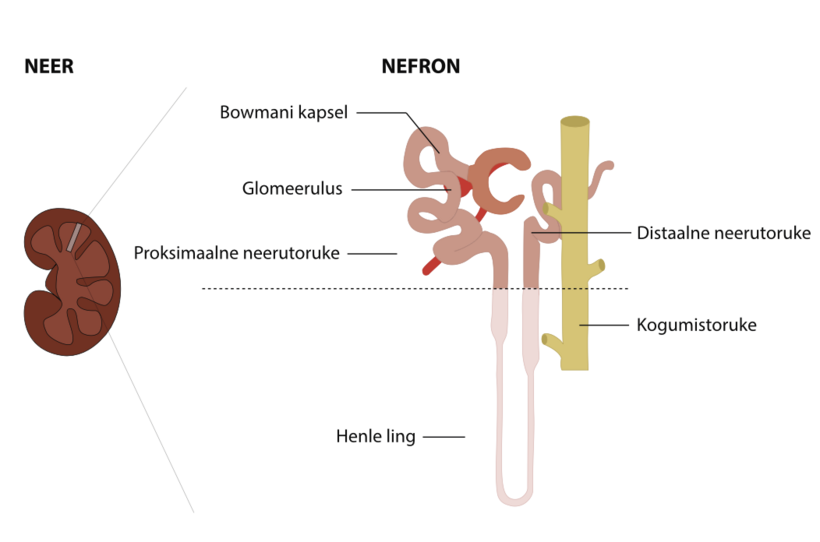
Neerude peamine ülesanne on eemaldada organismist liigne vesi ja jääkained, moodustades uriini. Igas neerus on keskmiselt umbes miljon nefronit, millest igaüks koosneb fi ltrist ehk neerupäsmakesest (glomeerulus) ja neerutorukesest (tuubulus). Glomeeruluses moodustub vere fi ltreerimise tulemusena esmasuriin (normaalses esmasuriinis ei ole vererakke ega suuri valke).
Tuubulites toimub glomeerulustes fi ltreeritud organismile vajalike ainete töötlemine – nt vesi, elektrolüüdid (naatrium, kloriid, kaalium, kaltsium, magneesium, fosfor ja paljud teised), glükoos, aminohapped ja valgud. Tuubulid reguleerivad ka happe-aluse homöostaasi. Need protsessid on vajalikud kehakemikaalide stabiilse tasakaalu säilitamiseks. Täiskasvanu neerud fi ltreerivad päevas umbes 150 liitrit vett, millest 99% imendub tuubulites tagasi, jättes umbes 1,5 liitrit lõplikku uriini. See on võimalik tänu sellele, et neerutuubulite kogupikkus on 80 km!
Tuubulid koosnevad järgmistest segmentidest: proksimaalne tuubul, Henle’i ling, distaalne tuubul ja kogumiskanal. Proksimaalsetes tuubulites toimub suurem osa reabsorptsioonist. Neerud toodavad ka hormoone, mis mõjutavad teiste organite funktsiooni (näiteks punaste vereliblede tootmist stimuleerivat hormooni). Teised neerude toodetud hormoonid aitavad reguleerida vererõhku ja kontrollida kaltsiumi ainevahetust.

Haiguse põhjuseks on ühe geeni CLCN5 või OCRL1 mutatsioon. Mõlemad neist geenidest asuvad sugukromosoomil X, mistõttu nende mutatsioonide ja haiguse pärimine on seotud sooga.
Meestel on igas keharakus üks X-kromosoom ja üks Y-kromosoom, naistel kaks X-kromosoomi.
Dent’i haiguse pärimine on retsessiivselt seotud X-kromosoomiga. See tähendab, et see esineb tavaliselt ainult meestel, kuna naistel on kaks X-kromosoomi, millest üks võib kompenseerida teise X-kromosoomi defekti. Haigus avaldub ainult siis, kui inimesel puudub vähemalt üks terve X-kromosoom.
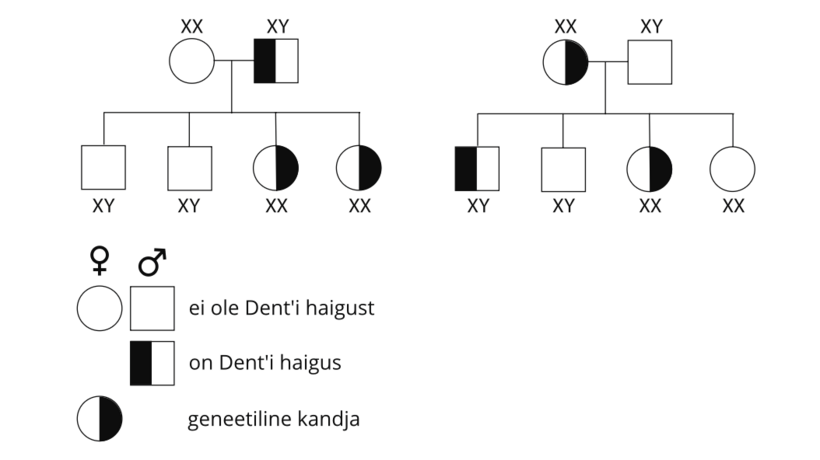
Poisid pärivad X-kromosoomi alati emalt ja Y-kromosoomi isalt – seega saavad Dent’i haiguse pärida ainult oma emalt. Kui ema on CLCN5 või OCRL1 mutatsiooni kandja, on 50% tõenäosus, et ta annab mutatsiooni edasi oma mõlemast soost lastele. Tüdrukud siiski ei haigestu, sest nad saavad isalt terve X-kromosoomi. Poistele jääb aga üks muteerunud X-kromosoom, sest nad pärivad isalt Y-kromosoomi.
Naisterahvast, kellel on haigestunud ja terve X-kromosoom, nimetatakse haiguse kandjaks, sest tal ei esine sümptomeid, kuid ta võib haiguse oma lastele edasi anda. Kuna vaja on ainult ühte aktiivset X-kromosoomi, on naistel üks kahest inaktiveeritud. See on juhuslik sündmus, mis toimub igas keharakus eraldi. Seega on naistel, kes on haiguse kandjad, mutatsiooniga ja mutatsioonita rakkude segu. Mõnel juhul võib naistel olla kasulik X-i inaktiveerimine, mille puhul on kahjustatud X-kromosoom enamikus rakkudes vaigistatud. Nendel naistel ei pruugi tekkida mingeid või ainult väga kergeid häire sümptomeid.
Teistel juhtudel võib naistel olla kahjulik X-inaktivatsioon, st intaktne X-kromosoom on enamikus rakkudes vaigistatud ning neil naistel võivad avalduda erinevad Dent’i haiguse sümptomid.
Äärmiselt harvadel juhtudel tekib mutatsioon juhuslikult ilma nähtava põhjuseta, kuna mõlemad vanemad ei kanna seda mutatsiooni. Seda nimetatakse „de novo mutatsiooniks“ ja selle põhjuseks on ühe vanema sugurakus (munarakk või spermatosoid) esinev või varase embrüogeneesi käigus viljastatud munarakkudes endas tekkiv mutatsioon.
Sellises olukorras avaldub muutus esimest korda sellel pereliikmel ja kõik teised tema vanemate järeltulijad ei ole mõjutatud. Siiski annab ta mutatsiooni edasi kõigile oma tütardele. Mutatsiooni päritolu (de novo vs. pärilik) kindlaksmääramine võib olla oluline edasise pereplaneerimise seisukohast (õdede-vendade riski hindamine), aga ka juhul, kui kroonilise neeruhaigusega pojale kaalutakse võimaliku neerudoonorina tema ema.
Inimkeha koosneb miljonitest rakkudest. Enamik rakke sisaldab täielikku geenikomplekti. Geenid on „elu retsept“ , milles on meie kasvu ja keha toimimist kontrollivad juhised. Need vastutavad paljude meie omaduste, näiteks silmade värvi või kehapikkuse eest. Geenimuutuse korral võib valkude tootmine olla defektne, ebaefektiivne või puududa. Sõltuvalt konkreetse valgu funktsioonist võib see mõjutada ühte või mitut organsüsteemi. Geenid koosnevad keemilisest ainest, mida nimetatakse DNA-ks, ja asuvad kromosoomideks nimetatud niitstruktuuride sees.
Igal inimesel on enamikus rakkudes 46 kromosoomi. Need on 22 paari autosoomseid kromosoomi ja 1 paar sugukromosoomi, st X ja Y. Kromosoomid pärandatakse vanematelt, 23 emalt ja 23 isalt, seega on igal inimesel 2 täielikku 23 kromosoomi komplekti ehk 23 „paari“. Kuna kromosoomid koosnevad geenidest, pärivad kõik inimesed enamiku geenide 2 koopiat, ühe koopia mõlemalt vanemalt. Sugukromosoomide puhul on olukord veidi teistsugune, kus meessugupoolel on üks X- ja Y-kromosoom ning naissugupoolel vastavalt kaks X-kromosoomi.
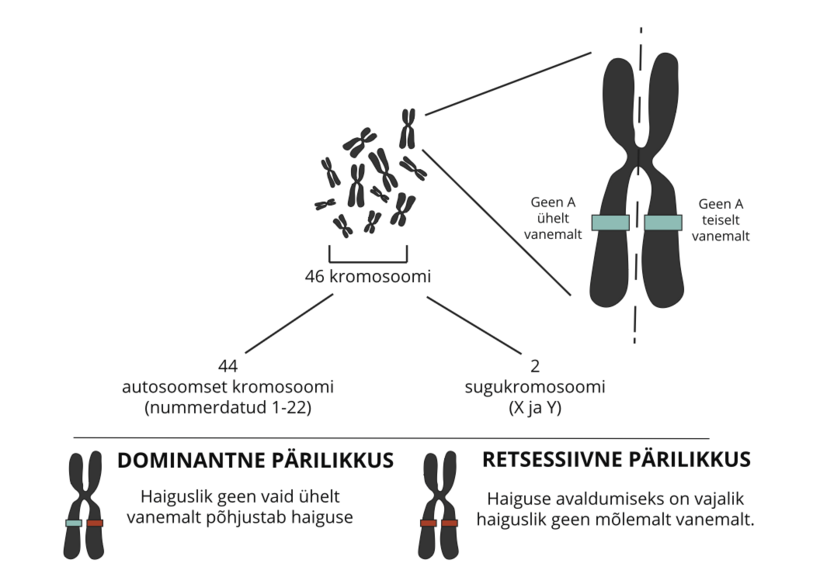
Seda tüüpi pärimine puudutab geene, mis asuvad autosomaalsetel kromosoomidel, st ei ole seotud sooga. Dominantse pärimise korral piisab haiguse sümptomite avaldumiseks ühest defektse geeni koopiast. Retsessiivse pärilikkuse korral takistab haiguse ilmnemist ühe geeni õige versiooni olemasolu, st haiguse ilmnemiseks on vaja kahte haiguslikku geeni.
X-liiteline pärilikkus tähendab, et geenid, mille mutatsioonid põhjustavad antud haigusüksust, asuvad X-kromosoomis. Need haigused erinevad autosoomsetest haigustest selle poolest, et need mõjutavad naisi ja mehi erinevalt. See on tingitud sellest, et mõlemal sugupoolel on erinev sugukromosoomide komplekt: Kui naistel on kaks X-kromosoomi ja seega iga geeni kaks versiooni X-kromosoomil, siis meestel on ainult üks X-kromosoom, st ainult üks versioon antud geenist. Seetõttu põhjustab defektne geen X-kromosoomil mehel haiguse, samas kui naistel võib teine X-kromosoom seda kompenseerida (X-ga seotud retsessiivne pärilikkus).
1., 2. ja 3. tüüpi Dent’i haigus
Seni on tuvastatud kahte tüüpi haigust. Mõlemad vormid on pärilikud retsessiivselt, kuid haigust põhjustavad mutatsioonid kahes geenis.
Haiguse põhjuseks on mutatsioon CLCN5 geenis, mis määrab kloriidikanali CLC-5 funktsiooni. CLC-5 mängib võtmerolli proksimaalsete tuubulite rakkude endosoomide hapestumisel - see on oluline madala molekulmassiga (LMW) valkude tagasiimendumisel uriinist. Tüüp 1on kõige levinum haiguse vorm ja moodustab umbes 65% kõigist Dent’i haiguse juhtudest. Selles geenis on palju erinevaid mutatsioone ja sümptomid võivad isegi sama mutatsiooni omavatel patsientidel olla väga erinevad.
Tüüp 2 esineb umbes 10-15% Dent’i haigusega patsientidest ja seda põhjustavad mutatsioonid OCRL1 geenis. Seda haigustüüpi iseloomustavad samad neerusümptomid, mis võivad esineda 1. tüüpi haiguse puhul, kuid lisaks võivad esineda sellised sümptomid nagu kerge vaimne mahajäämus, kerge katarakt ning samuti võib täheldada lühikest kasvu. OCRL geen kodeerib valku, mis osaleb proksimaalsete neerutorukeste rakkude rakusiseses transpordis. See valk osaleb ka paljudes teistes protsessides organismis. Mõned OCRL-geeni mutatsioonid põhjustavad 2. tüüpi Dent’i haigust, kuid teised palju raskemat haigust, mida nimetatakse Lowe’i sündroomiks (vt allpool).
See puudutab ülejäänud patsiente, kellel on Dent’i haiguse tunnused (25-35%), kuid kellel ei ole suudetud tõestada ühtegi eespool nimetatud mutatsiooni. On tõenäoline, et seda haigust põhjustavad teised geenid, mis on seni teadmata.
Dent’i haigustkirjeldasid esmakordselt Charles Enrique Dent ja M. Friedman 1964. aastal, kui nad teatasid kahest omavahel mitteseotud Briti poisist, kellel esines rahhiit ja neerutorukeste kahjustus, mida iseloomustas hüperkaltsuuria, hüperfosfatuuuria, proteinuuria ja ebanormaalselt suur aminohapete hulk uriinis. Haiguse nimi anti 30 aastat hiljem, kui nefroloog Oliver Wrong kirjeldas haigust põhjalikumalt ja otsustas nimetada haiguse oma mentori järgi. Haigus esineb peaaegu eranditult meestel ja sümptomid võivad ilmneda juba varases lapsepõlves.
Dent’i haiguse raskusaste võib olla väga erinev isegi haigestunud pereliikmete puhul. Haigestunutel ei pruugi olla kõiki allpool kirjeldatud sümptomeid. Üksiku haigusjuhu korral tuleb hinnata haigustunnuseid ning nende ravi ja haiguse prognoosi hinnata juhupõhiselt. Haiguse juhuslik diagnoosimine asümptomaatilises faasis on võimalik, kuid enamasti diagnoositakse see 30-50-aastaselt teadmata etioloogiaga kaugelearenenud kroonilise neeruhaiguse korral. Dent’i haiguse mutatsioone kandvatel naistel võib esineda kerget väikemolekulaarset proteinuuriat ja hüperkaltsuuriat, kuid neerukivid ja neerupuudulikkus on haruldased.
Madala molekulmassiga proteinuuria (low molecular weight ehk LMW proteinuuria)
Tavaliselt on tegemist enam kui viiekordselt suurenenud beeta-2-mikroglobuliini eritumisega uriini!
Dent’i haigusega patsientidel on uriinis suurenenud valgusisaldus. See on ainus haiguse sümptom, mis laboratoorselt püsib. Dent’i haiguse puhul täheldatavat proteinuuria tüüpi nimetatakse madala molekulmassiga proteinuuriaks (LMW-proteinuuria).
LMW-valgud on väikesed valgud, mis fi ltreeritakse neerude poolt, kuid imenduvad tagasi ja lagundatakse proksimaalsetes tuubulites, nii et neid ei saa tervete inimeste uriinis tuvastada. LMW-valkude mõõtmiseks on vaja spetsiaalseid teste ja need võivad jääda rutiinsete uriinianalüüside käigus tähelepanuta.
Diagnostikas kasutatavad LMW-valgud on näiteks beeta-2-mikroglobuliin, alfa-1-mikroglobuliin ja retinooli siduv valk. LMW-valgu esinemine uriinis viitab proksimaalse tuubuli häiritud funktsioonile. Dent’i haiguse korral on beeta-2-mikroglobuliini kontsentratsioon uriinis vähemalt viis korda kõrgem kui normi ülemine piir. Madalamolekulaarse proteinuuria kvantitatiivseks hindamiseks võib teha 24-tunni uriini kogumise või hinnata beeta-2-mikroglobuliini (kui LMW-valgu näide) ja kreatiniini suhet juhuslikus uriiniportsjonis. Kuna valkude esinemine uriinis on pigem neerupäsmakese kui neerutorukestehaigestumise tunnus, võivad arstid Dent’i haiguse esialgu ekslikult diagnoosida glomerulonefriidi (neerupäsmakese põletik) või nefrootilise sündroomina (valkude massiline lekkimine läbi glomerulaarfi ltrite). Uriini üksikasjalikum analüüs näitab, et Dent’i haigusega patsientide uriinis on ülekaalus LMW-valgud.
Fanconi sündroom ja madal seerumi fosfaatide tase
Madala molekulmassiga valkude vähenenud tagasiimendumine proksimaalsetes neerutuubulites kaasneb sageli teiste ainete, nagu fosfaatide, kaaliumi, aminohapete või bikarbonaatide puuduliku imendumisega. Sõltuvalt mõjutatud ainete osakaalust nimetatakse seda osaliseks või täielikuks neeru Fanconi sündroomiks.
Täielik Fanconi sündroom (De Toni-Debré-Fanconi sündroom) - defekt, mis mõjutab kõiki proksimaalsete tubulite funktsioone, põhjustades aminohapete, glükoosi, fosfaatide, kusihappe, tsitraadi, väikese molekulmassiga valkude, magneesiumi, kaaliumi, kaltsiumi, bikarbonaadi ja vee kaotust.
Osaline Fanconi sündroom viitab ainult mõne eespool nimetatud komponendi kaotusele. Enamikul Dent’i haigusega patsientidel ei ole kõik proksimaalse tuubuli funktsioonid kahjustatud. Pidev rohke eritumine neerude kaudu põhjustab ainete kontsentratsioonide vähenemise veres: fosfaadikaotus põhjustab hüpofosfateemia, kaaliumikaotus hüpokaleemia, bikarbonaatide kaotus neeru tubulaarse atsidoosi. Aminohapete kadu ei põhjusta mingeid metaboolseid tagajärgi. Kuna fosfaat on vajalik luu moodustamiseks, võib hüpofosfateemia põhjustada rahhiiti või osteomalaatsiat (vt sõnastikus), mis (erinevalt teistest rahhiidivormidest) ei reageeri suurtele D-vitamiini annustele. Neeru tubulaarne atsidoos süvendab luukahjustust ning tekivad muutused luudes ja kasv võib olla. Hüpokaleemia võib põhjustada lihasnõrkust ja kahjustada vee imendumist ning selle tagajärjeks onsuurenenud uriini tootmine (polüuuria) ja janu (polüdipsia) ning kõrge vedelikupuuduse risk.
Hüperkaltsuuria – kaltsiumi suurenenud eritumine uriiniga
Hüperkaltsuuriat, nagu ka madala molekulmassiga valgueritust, saab tuvastada ainult laboratoorsete testidega. Silmaga uriini hindamisel seda ei näe, ehkki samal ajal võib kaasneda ka veri uriinis ehk hematuuria. Dent’i haiguse hüperkaltsuuria põhjus ei ole veel täielikult teada. Võimalik mehhanism on kõrvalkilpnäärmehormooni tagasihaarde ebaõnnestumine. Kõrvalkilpnäärmehormoon madalmoleklaarne valk, mõjutab kaltsiumi eritumist Samutivõib kaasneda D-vitamiini siduvate valkude kaotus. Teine mehhanism võib olla Dent’i haigusele iseloomulikust metaboolsest atsidoosist põhjustatud suurenenud kaltsiumi vabanemine luu resorptsioonist. Kaltsiumi eritumise hindamiseks uriiniga soovitatakse 24h uriini kogumist. Kui see ei ole võimalik, näiteks kui laps kannab veel mähkmeid, võib määrata kaltsiumi ja kreatiniini kontsentratsiooni suhet uriini osas, kuigi see on vähem täpne.
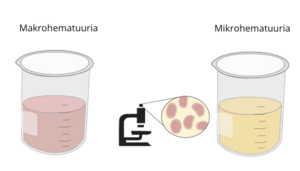
Neerude kaltsifi katsioonid (nefrokaltsinoos) või neerukivid (nefrolitiaas)
Suurenenud kaltsiumikontsentratsioon uriinis põhjustab kristalliseerumist ja kaltsifi katsioonide teket neerukoes (nefrokaltsinoos) ning neerukivide teket (nefrolitiaas). Neerukaltsifi katsioonide ja neerukivide teket saab tuvastada ultraheliuuringuga. Mõnikord võib esimeseks sümptomiks olla “mikrohematuuria” – veri uriinis, mida on võimalik näha ainult mikroskoopilisel uuringul. See võib olla esimene neerukividele viitav sümptom. Neerukivid võivad põhjustada ka valulikku urineerimist (düsuuriat), sagedast urineerimissoovi, kõhuvalu (neerukoolik), urineerimistakistust või korduvaid kuseteede põletikke.
Krooniline neeruhaigus ja neerupuudulikkus
Haiguse progresseerumine võib põhjustada kroonilist neeruhaigust (CKD) koos neerufunktsiooni progresseeruva vähenemisega. Väga kaugelearenenud kroonilise neeruhaigusega seotud sümptomite hulka kuuluvad söögiisu kadumine, tahtmatu kehakaalu langus, väsimus ja aneemia. Mõnel juhul, mõnikord juba 30-50-aastaselt, võib haigestunud inimestel tekkida lõppstaadiumi neerupuudulikkus ja nad vajavad dialüüsi või neerusiirdamist.
Muud sümptomid
Mõnedel Dent’i haigusega inimestel võivad tekkida luuhaigused, nagu luupehmenemine (osteomalaatsia) ja hüpofosfateemiline rahhiit (seisund, mille põhjuseks on fosfaatide vähenenud transport ja muutunud D-vitamiini ainevahetus neerudes).
Dent’i haigusega lastel võib kasvutempo olla normaalsest aeglasem, mille tulemuseks on sageli kerge lühike kasv. Lastel võivad tekkida ka luuvalu ja kõndimisraskused. Luu kõrvalekallete tõttu võib nii lastel kui ka täiskasvanutel olla suurenenud luumurdude risk.
Mõnedel Dent’i haigusega inimestel on A-vitamiini puudus, mis võib põhjustada öise nägemise halvenemist ja kuivsilmust. Vitamiin A puudus on sel juhul põhjustatud madala molekulaarse valgu sidumise retinooli kaotusest uriiniga. Sümptomeid saab korrigeerida vitamiinasendusraviga.
Mõnedel 2. Tüüpi Dent’i haiguse all kannatavatel inimestel võivad esineda muud sümptomid, sealhulgas kerge vaimne alaareng, vähenenud lihastoonus ja sellega seotud hilinenud motoorne areng, samuti silmaläätse hägustumine (katarakt), tavaliselt ei põhjusta nägemishäireid.

Madala molekulmassiga proteinuuria koos hüperkaltsuuria või nefrokaltsinoosiga või neerukividega poisil/mehe puhul võivad olla ainsad haiguse sümptomid ja peaksid ajendama edasist diagnostikat.
Dent’i haiguse kliiniline diagnoosimine põhineb iseloomulike sümptomite tuvastamisel, patsiendi ja perekonna üksikasjaliku anamneesi hindamisel, põhjalikul kliinilisel hindamisel ja mitmesugustel spetsiaalsetel testidel.
Kuid haiguse sümptomite mitmekesise iseloomu tõttu tuleks Dent’i haigust kaaluda ka meestel, kellel on:
- glomerulaarse haiguse sümptomid (steroidresistentne nefrootiline sündroom)
- neerude tubulaarsed häired/Fanconi sündroom
- idiopaatiline nefrolitiaas ehk teadmata põhjusega neerukivitõbi
- täpsustamata põhjusega krooniline neeruhaigus
Molekulaargeneetiliste testidega saab tuvastada mutatsioonid kahes geenis, mis teadaolevalt põhjustavad Dent’i haigust, kuid kuid kui kliiniline diagnoos on võimalik (näiteks madala molekulmassiga proteinuuria ja hüperkaltsuuria meestel), ei ole see alati vajalik. Teisalt, on molekulaargeneetilised testid soovitavad, eristamaks seda teistest nefrokaltsinoosi ja kroonilise neeruhaiguse geneetilistest põhjustest. Umbes kolmandikul Dent’i haiguse tüüpilise pildiga meestest ei leita mutatsiooni kahes Dent’i geenis.
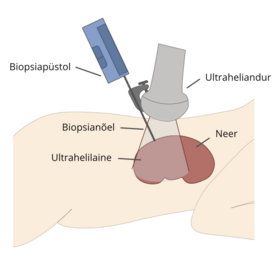
Neerubiopsia (väikese neerukoe proovi võtmine mikroskoopiliseks hindamiseks) tehakse sageli märkimisväärse proteinuuria või hematuuria korral diagnoosi kinnitamiseks. Mõnikord teostatakse neerubiopsia Dent’i haigusega patsientidele enne põhidiagnoosini jõudmist. Biopsia tulemused on mittespetsiifi lised ja näitavad fokaalset glomerulaarskleroosi (FSGS) (vt sõnastik), interstitsiaalset fi broosi (armistumist) ja nefrokaltsinoosi.
Neerukoebiopsia ei ole Dent’i haiguse diagnoosimiseks vajalik ja võib olla isegi eksitav.
Kas Dent’I haigus võib olla segi aetud mõne muu haigusega?
On mitmeid haruldasi geneetilisi häireid, mida iseloomustab neeru- või kuseteede kivide teke lapsepõlves ja mis sarnanevad Dent’i haigusega. Selliste häirete hulka kuuluvad primaarne hüperoksaluuria, perekondlik hüperkaltsuuria-hüpomagneseemia-nefrokaltsinoos (Michelis-Castrillo sündroom), adeniinfosforibosüültransferaasi (APRT) puudulikkus ja tsüstinuuria.
Primaarne hüperoksaluuria (PH) | on rühm haruldasi geneetilisi ainevahetushäireid, mida iseloomustab oksalaadina tuntud aine kuhjumine neerudes ja teistes organitesüsteemides, sest haigetel puudub ensüüm, mis tavaliselt takistab oksalaadi kuhjumist. |
Perekondlik hüperkaltsuuria - hüpomagneseemia-nefrokaltsinoos (Michelis-Castrillo sündroom, FHHNC) | on haruldane geneetiline haigus, mis pärandub autosoom-retsessiivselt ja mille puhul esineb magneesiumi ja kaltsiumi liigne eritamine uriiniga. Haiguse põhijooned on hüpomagneseemia (vereseerumi madal magneesiumi väärtus), hüperkaltsuuria (vereseerumi kõrge kaltsiumi väärtusja nefrokaltsinoos ning patsientidel esineb kliiniliselt polüuuria/polüdipsia ja D-vitamiini suhtes resistentne rahhiit. FHHNC tuleneb CLDN16 või CLDN19 geenide mutatsioonidest. CLDN16 ja CLDN19 kodeerivad vastavalt claudin-16 ja claudin-19 valke, mis ekspresseeruvad Henle’i lingu ülenevas osas ja moodustavad magneesiumi ja kaltsiumi paratsellulaarse tagasiimendumise jaoks olulise kompleksi. Nendel patsientidel ei esine hüpokaleemiat ega soolade kaotust. CLDN19 mutatsioonidega patsientidel esinevad rasked silmade kõrvalekalded, nagu lühinägelikkus, nüstagm ja makulaarne koloboom (silma kollatähni kaasasündinud puudulikkus). |
Adeniinfosforibosüültransferaasi (APRT) puudulikkusele | on iseloomulik 2,8-dihüdroksüadeniini (DHA) ülemäärane tootmine ja eritumine neerude kaudu, mis põhjustab neerukivide teket ja kristallidest põhjustatud neerukahjustust (st DHA-kristallide nefropaatiat), mis põhjustab ägeda neerukahjustuse episoode ja süveneva kroonilist neeruhaigust. |
Tsüstinuuria | on pärilik autosoom-retsessiivne haigus, mida iseloomustab aminohappe tsüstiini kõrge kontsentratsioon uriinis, mistõttu moodustuvad neerudes ja kuseteedes tsüstiinkivid. |
Fanconi Sündroom | Mitmeid pärilikke häireid iseloomustab Fanconi sündroom ja seetõttu tuleks seda Dent’i haiguse eristamisel samuti arvesse võtta. Näidetena võib tuua sellised geneetilised häired nagu tsüstinoos, pärilik fruktoositalumatus, galaktoseemia, türosineemia, Wilsoni tõbi ja mitmesugused glükogeeni ladustamise haigused. Neeru Fanconi sündroom võib tekkida ka elu jooksul teatud ravimite (nt valproaat, deferasirox, tsisplatiin, ifosfamiid) kõrvaltoimena või olla sekundaarne teatud neeruhaiguste, vähkkasvajate, nt hulgimüeloomi, Sjögreni sündroomi või hüperparatüreoidismi tagajärjel. Lastel võib kokkupuude raskemetallidega põhjustada ka Fanconi sündroomi. |
Lowe’i sündroom | on haruldane geneetiline häire, mida iseloomustavad nägemishäired, sealhulgas silmaläätsede hägustumine (katarakt), mis esineb juba sünnihetkel, neeruprobleemid, mis tekivad tavaliselt esimesel eluaastal, ja intellektuaalne mahajäämus. Haiguse konkreetsed sümptomid ja raskusaste võivad olla inimeseti väga erinevad. Lowe’i sündroom pärandub X-liiteliselt. Lowe’i sündroomi põhjustavad mutatsioonid samas geenis (OCRL1), mis põhjustavad 2. tüüpi Dent’i haigust. Häire avaldub täielikult ainult meestel. |
Proteinuuria ehk ülemäärane valgueritus uriinis esineb ka glomerulonefriitide korral. Neid haigusi ravitakse sageli kortikosteroididega, kuid Dent’i haiguse korral on see tulemusetu, sest Dent’i haigus ei ole neerupäsmakese põletik vaid põhjustatud geenimuutusest. Asjaolu, et glükokortikosteroidid ei aita, võib panna arsti liigitama Denti haigust “steroidresistentseks nefrootiliseks sündroomiks” enne õige diagnoosi püstitamist. Eriti eksitav võib olla see, kui tehakse neerubiopsia ja neerukoe uurimise tulemusel nähakse fokaalset glomerulaarskleroosi.

Kes osaleb minu või minu lapse ravis?
Dent’i haiguse ravi peaks suunama nefroloog ja selle eesmärk on tavaliselt sümptomite vähendamine neerukivide teket pärssivate ravimite ning elektrolüütide ja ainevahetuse häireid korrigeerivate toidulisandite võtmise abil.
Ravi võib nõuda eriarstide meeskonna koordineeritud koostööd: lastearstid, nefroloogid ja uroloogid, dietoloogid ja muud tervishoiutöötajad.
Ravi koosneb järgmistest meetmetest:
- Kaltsiumkivide ladestumise vähendamine(profülaktiline ravi, kivide tekke vältimine, nn metafülaktika)
- Kivide eemaldamine kuseteedest (sümptomaatiline ravi)
- Haiguse tagajärgede ravi, sealhulgas elektrolüütide häirete korrigeerimine, samuti progresseeruva kroonilise neeruhaiguseja selle tüsistuste ravi
Piisav vedeliku tarbimine > 3 liitrit/m2 kehapinna kohta
Piisav, antud juhul keskmisest suurem vedelikutarbimine on oluline sekkumine, mis vähendab kaltsiumi ladestumist neerudes. Arvatakse, et soovituslikust vähema vedelikutarbimise korral väheneb ka teiste ennetusmeetodite tõhusus. Igapäevase vedelikuvajaduse hindamiseks võib kehapindala (BSA) arvutada internetis kättesaadavate kalkulaatorite või valemi abil:
BSA=√(kehakaal (kg) x pikkus (cm)/3600)
Ärge unustage, et kõhulahtisuse, palaviku, oksendamise või kuumas kliimas viibimise korral vedelikuvajadus suureneb. Seetõttu peaks suurenenud vedelikukaotus või vähenenud vedelikutarbimine nõudma viivitamatult arstiabi, et vältida haiguse järsku progresseerumist. Kui suukaudne vedeliku manustamine ei ole võimalik, võib täiendav vedeliku manustamine olla vajalik isegi veenisiseselt. Patsiendid, nende hooldajad ja õpetajad peavad olema teadlikud haigusest ja suurest vedelikutarbimise vajadusest.
Pange tähele:
- Hoidke alati kaasas dokument, mis tõendab teie/ teie lapse haigust
- Igapäevane vedeliku tarbimine peaks ületama 2,5-3 liitrit/m² kehapinna kohta
- Veenduge, et teil on endaga alati piisavalt palju vedelikku kaasas
- Jooge vet regulaarsete ajavahemike järel kogu päeva jooksul, ka siis, kui teil ei ole janutunnet
- Planeerige sagedased tualetiskäigud
Seadistage vajadusel oma mobiiltelefoni meeldetuletused, et meenutada vee joomist, samuti võite kasutada spetsiaalseid rakendusi, et registreerida, kui palju vett te joote.
Toitumine
Söögisoola tarbimise piiramine vähendab uriiniga erituva kaltsiumi kogust ja seega ka neerukivide tekkimise riski.
Ravimite kasutamine
Kaaliumi ja fosfaadi lisamine
Kuna Dent’i haigusele on iseloomulikud oluliste mineraalainete, näiteks kaaliumi kaotus neerutorukestes, on vajalik nende asendus. Patsientidele, kellel on madal fosfaatide sisaldus (hüpofosfateemia) ja rahhiit, määratakse fosfaadilisandeid. Fosfaatide lisamine võib vähendada hüperkaltsuuriat isegi hüpofosfateemia puudumisel. Kaltsiumikristallide ladestumist vähendavad ravimid (koos vedeliku tarbimisega) võivad veelgi vähendada kivide moodustumise riski. Parimad tulemused saavutatakse, kui ravimeid võetakse päeva jooksul regulaarsete ajavahemike järel.
Kaaliumtsitraat moodustab uriinis kaltsiumiga lahustuvaid komplekse, vähendades seeläbi kaltsiumi kättesaadavust kristallide moodustumiseks. Tsitraat metaboliseerub maksas bikarbonaadiks ja viib vere ja uriini leeliselisemaks (vere ja uriini kõrgem pH). Sellistes tingimustes imendub neerutorudes vähem tsitraati tagasi ja seda rohkem eritub uriiniga. Tsitraat seob kaltsiumi ja vähendab kaltsiumi kättesaadavust kivide moodustamiseks. Leeliseliste tsitraatide annust reguleeritakse individuaalselt uriini pH-testi tulemuse alusel, mida kontrollitakse pH-indikaatorpaberi abil. Siht-pH väärtused jäävad vahemikku 6,2-7,4. Krooniline ravi tsitraadiga võib edasi lükata neeruhaiguse progresseerumist ja isegi ennetada kivide teket.
Tiasiiddiureetikume määratakse Dent’i haiguse korral sageli hüperkaltsuuria vähendamiseks, kuid nende kasutamist piiravad kõrvaltoimed nagu hüpovoleemia (kehavedelike vähenemine, veekaotus) ja hüpokaleemia (seerumi kaaliumisisalduse vähenemine). Seetõttu on oluline patsiendi hoolikas jälgimine ravi ajal ning eriti ettavaadlik tuleb olla vähenenud vedelikutarbimise või suurenenud vedelikukaotuse korral.
Mõned nefroloogid määravad AKE-inhibiitorid, et vähendada proteinuuriat, sest need ravimid vähendavad fi ltratsioonirõhku neerupäsmakestes ning seega glomerulaarfi ltreid läbivate valkude hulka. Selline ravi on vasturääkiv, sest Dent’i haigusepuhul on probleemiks valkude vähenenud omastamine neerutorukeses, mitte suurenenud fi ltratsioon neerupäsmakeses. Siiani ei ole tehtud ühtegi uuringut, et kontrollida, kas AKE-inhibiitorist on Denti haiguse puhul kasu.
on väheselt invasiivne meetod kivide eemaldamiseks kuseteedest, mille puhul pääseb neerule ligi kusejuha kaudu ja kivid purustatakse laseri abil. Üldnarkoosis viiakse endoskoop kusiti kaudu põide ja kanüülimise järel visualiseeritakse kivi kusejuhas. Sõltuvalt kivi suurusest on purustatud kiviosakesed piisavalt väikesed, et need läheksid iseeneslikult välja või neid saab endoskoobi abil eemaldada.
Isegi tänapäeval diagnoositakse Dent’i haigus sageli alles siis, kui neerufunktsioon on juba oluliselt häiritud. Sellistel juhtudel on vajalik kroonilise neeruhaiguse ja selle tüsistuste ravi.
Neerufunktsiooni säilitamine nii kaua kui võimalik
Et säilitada neerufunktsiooni võimalikult kaua, on oluline pöörata erilist tähelepanu teguritele, mis võivad neerufunktsiooni kahjustada. Sel põhjusel on soovitatav:
Vältida ravimeid, mis võivad kahjustada neerusid, näiteks mittesteroidseid põletikuvastaseid ravimeid („MSPVA“) - nagu ibuprofeen, naprokseen jt, mis on paljudes apteekides ilma retseptita saadaval. On mitmeid teisi ravimeid, mida tuleks vältida – teie raviarst on sellest teadlik.
Kontrastainete vältimine või väga ettevaatlik kasutamine radioloogilistel uuringutel, näiteks kompuutertomograafi a (KT).
Olenemata sellest, millised on muud tervisehäired, teavitage alati oma arsti Dent’i haiguse diagnoosist, et ta oskaks teid paremini jälgida ning saaks vajadusel ravi kohandada.
Neeru siirdamine
Lõppstaadiumis neeruhaiguse korral, kui neerud ei suuda eritada piisavalt ainevahetusjääke, et hoida keha elus, alustatakse dialüüsi või tehakse neerusiirdamine.
Dialüüs
Dialüüs on protseduur, mille puhul neerude põhifunktsiooni – st vee ja ainevahetuse jääkainete eemaldamine teostatakse, kasutates vere jääkainetest puhastamiseks spetsiaalset fi ltrit (hemodialüüs) või kõhukelmeõõnt (peritoneaaldialüüs):
Hemodialüüsi puhul pumbatakse veri läbi fi ltri, kus see puhastatakse ja eemaldatakse liigne vedelik. Seda meetodit tehakse tavaliselt haiglas, nt 4 korda nädalas mitu tundi.
Peritoneaaldialüüs hõlmab dialüüsivedeliku korduvat manustamist ja eemaldamist kõhuõõnde või sealt välja kateetri abil. Seda meetodit saab teha kodus spetsiaalse seadme abil, tavaliselt öösel, kui patsient magab.
Seni ei ole veel välja töötatud ühtegi haiguse põhjuslikku ravi, samuti ei ole esinevate sümptomite mitmekesisuse tõttu standardset ravi. Dent’i haiguse harvaesinemise tõttu puuduvad kliinilised uuringud suure patsientide rühmaga, mis tõestaksid näiteks tsitraadi või tiasiidide tõhusust. Siiski on katsed Denti haiguse loommudelitel andnud olulisi teadmisi. Dent’i haiguse raviks tehtavad geeniteraapiauuringud on käimas. Eksperimentaalsed andmed näitavad, et 1. tüüpi Dent’i haiguse puhul muteerunud valgu CLC5 defektne funktsioon võib olla päästetav nn väikemolekulaarsete ravimeetoditega.
Dent’i haigusega patsientidel on suur oht vedelikupuuduseks ehk dehüdratsiooniks ja palaviku, kõhulahtisuse/oksendamise ja teiste ägedate haigestumiste korral võivad patsiendid vajada vedelikasendusravi ja elektrolüütide jälgimist haiglas. Kui patsientidel on neerukivid, võib see põhjustada kuseteede obstruktsiooni, mis nõuab kiiret uroloogilist sekkumist, et vältida neerufunktsiooni halvenemist.
Teavitage kirurgi/anestesioloogi diagnoosist „Dent’i haigus“. Operatsiooni eelselt on kõigil Dent’i haigusega patsientidel hinnata neerufunktsiooni ja vere elektrolüütide (eelkõige kaaliumi) taset vereseerumis. Võimaluse korral võtke enne operatsiooni või muid meditsiinilisi sekkumisi ühendust teid/ teie last jälgiva (laste)nefroloogiga, et arutada teie/ teie lapse erinõudeid/vajadusi või piiranguid.
Ettearvamatu kulg ja neerufunktsiooni äkilise halvenemise risk on suur psühholoogiline koormus haigestunud patsientidele ja nende perekondadele. Enamik patsiente, sealhulgas nende vanemad, vajavad psühholoogilist tuge.

Ehk millised on ravi pikaajalised tulemused?
Dent’i haigusega patsientide varasemate vaatluste põhjal avaldub 30-80% Dent’i haigusega meestest 30-50 aasta vanuselt krooniline neeruhaigus raskes raskusastmes. Haiguse harvaesineva iseloomu tõttu puuduvad andmed, milles hinnatakse varase diagnoosimise mõju edasisele prognoosile. Tundub siiski, et õigeaegne sekkumine vähendab neerudes kaltsiumiladestuste ja kivide teket ning nii võib aeglustada kroonilise neeruhaiguse süvenemist.

Kuidas ja kust saan ma rohkem abi?
Paljudes riikides on olemas Dent’i haigusega patsientide tugirühmad. Dent’i haigus on haruldane haigus ja seetõttu ei tea me sellest kõike, kuid teabe ja kogemuste vahetamine võib olla patsientidele ja nende peredele väga kasulik. Patsientide tugirühmad ja sihtasutused korraldavad kohtumisi, loenguid, aga veedavad ka koos aega ühistel suve/talvepäevadel.
Palun külastage Dent'i haiguse patsiendirühma siin.

AKE-inhibiitorid | ravimrühm, mis vähendab filtratsioonirõhku neerudes ja on glomerulaarhaiguste ravi alustala |
Aminoatsiduuria | ebanormaalselt suur aminohapete kogus uriinis |
| Kreatiniin | veres ringlev ainevahetusprodukt, mida kasutatakse neerufunktsiooni hindamiseks, sest seda fi ltreeritakse neerude poolt ja eritatakse uriiniga. Mida kõrgem on kreatiniini kontsentratsioon veres, seda halvem on neerufunktsioon |
Krooniline neeruhaigus (CKD ehk chronic kidney disease) | progresseeruv ja pöördumatu neerukahjustus, mis võib kuude või aastate jooksul viia lõppstaadiumi neeruhaiguseni |
| Dialyüüs | meetod ainevahetuse jääkainete ja liigse vedeliku eemaldamiseks verest. On olemas kaks peamist dialüüsi tüüpi: hemodialüüs ja peritoneaaldialüüs. Hemodialüüsi puhul kasutatakse vere puhastamiseks spetsiaalset masinat, mille fi ltra abil veri jääkainetest puhastatakse. Peritoneaaldialüüsi korral manustatakse dialüüsivedelik spetsiaalse vooliku abil kõhuõõnde ning fi ltrina toimib kõhukelme |
Lõppstaadiumi neeruhaigus (ESKD ehk end stage kidney disease) | neeruhaiguse kõige raskem vorm, kui neerud on lakanud töötamast (võivad siiski veel toota väga halva kvaliteediga uriini), tavaliselt on vajalik neeruasendusravi (dialüüs või neeru siirdamine) |
| Fanconi sündroom | sümptomite kogum, mille põhjuseks on proksimaalse neerutorukese defekt, mis põhjustab häireid aminohapete , glükoosi, fosfaadi, kusihappe, tsitraadi, väikeste valkude, magneesiumi, kaaliumi, kaltsiumi, bikarbonaadi ja vee tagasiimendumises |
Fokaal-segmentaalne glomeruloskleroos (FSGS) | seisund, mille puhul neerupäsmakestes tekkinud armkoe tõttu kujuneb neerukahjustus, mis võib süveneda lõppstaadiumi neerupuudulikkuseni. FSGS tunnuseks on tavaliselt suur valgukogus uriinis |
| Geen | geneetiline üksus, mis sisaldab juhiseid („retsepti“), kuidas iga valku organismis toota |
Glomeerulid | neerupäsmakesed, nefroni struktuuriüksused, milles toimub vere fi ltreerimine esmasuriiniks- Igas neerus on 250 000 kuni 1 miljonit glomeerulust |
(eGFR) hinnanguline glomerulaarfi ltratsiooni kiirus | arvutuslik verest jääkainete fi ltreerimise kiirus neerus. eGFR on tavaliselt suurem kui 90 ml/min/1,73m2 ning madalam väärtus viitab neerufunktsiooni kahjustusele. Väärtus alla 30 ml/min/1,73m2 vastab raskele neerupuudulikkusele, umbes 10 ml/min/1,73m2 juures on vajalik neeruasendusravi |
Neerusiirdamine | operatsioon, mille käigus kroonilise neeruhaiguse lõppstaadiumis olevale inimesele opereeritakse funktsioneeriv neer |
Lowe’i sündroom | okulo-tserebro-renaalne sündroom, mis on põhjustatud mutatsioonidest samas geenis (OCRL1), mis põhjustab Dent’i haiguse 2. tüübi, on samuti X-liiteline retsessiivselt pärinev haigus ning avaldub ainult meestel |
| Makrohematuuria | verikusesus, vererakkude esinemine uriinis |
| Mikrohematuuria | väikese hulga punaste vereliblede (erütrotsüütide) esinemine uriinis, mil uriini värvus on normaalne ja punavererakudon nähtavad ainult mikroskoopilisel uurimisel |
Nefrokaltsinoos | arvukate väikeste lubjastumiste ehk kaltsifi katsioonide esinemine neerukoes, mis peegeldavad kaltsiumkristallide ladestumist. Need muutused on kergesti nähtavad ultraheliuuringuga. Nefrokaltsinoos võib soodustada neerukivide teket |
| Nefron | on neeru põhiline funktsionaalne ja struktuuriline üksus, see koosneb kahest osast: neerupäsmakesest ehk glomeerulusest ja neerutorukesest ehk neerutuubulist |
Nefrootiline sündroom | seisund, mille puhul neerupäsmakestest lekib uriini valke ja seetõttu tekib veres valkude väike osakaal, organismis peetub vedelik, sageli on nähtavaks sümptomiks tursed silmade ümbruses ja jalgadel |
Osteomalaatsia | luude pehmenemine, mis on põhjustatud luuainevahetuse häiretest - tingitud peamiselt fosfaadi, kaltsiumi ja D-vitamiini ebapiisavast varustamisest. võib olla põhjustatud ka suurenenud kaltsiumi eraldumisest luudest |
Polüdipsia | liigne veejoomine, võib olla mitmete haiguste sümptom, mis põhjustavad veekaotust ja mille tulemuseks on liigne janutunne |
Polüuuria | ebanormaalselt suur uriinitoodang (nt täiskasvanutel üle 3 liitri päevas) |
Neerutuubul | nefroni osa, kus toimub neerupäsmakesest pärit esmase uriini molekulide eritamine ja tagasiimendumine Tuubulites töödeldakse ööpäevas umbes 150 liitrit esmast uriini, mille tulemusel eritub põiest umbes 1,5 liitrit uriini. Tuubulus koosneb mitmest osast: proksimaalne tuubulus, Henle’i ling, distaalne tuubulus ja kogumistoruke |
Rahhiit | kaltsium-fosfaat ainevahetuse häiretest põhjustatud luude väärarengud |
Tubulopaatiad | harvaesinevad neeruhaigused, mille puhul on neerutorukeste funktsioon häiritud, samas kui neerupäsmakesed toimivad normaalselt |
Urolitiaas (nefrolitiaas) | kivide moodustumine kuseteedes (neerudes) |
Gianesello L, Del Prete D, Anglani F, Calò LA.Genetics and phenotypic heterogeneity of Dent disease: the dark side of the moon.Hum Genet.2021;140(3):401-421. doi: 10.1007/s00439-020-02219-2. Epub 2020 Aug 29. PMID: 32860533
Anglani F, Gianesello L, Beara-Lasic L, Lieske J. Dent disease: a window into calcium and phosphate transport. J Cell Mol Med. 2019;23:7132–7142. doi:10.1111/jcmm.14590.-DOI-PMC–PubMed
van Berkel Y, Ludwig M, van Wijk J, Bökenkamp A. Proteinuria in Dent disease: a review of the literature. Pediatr Nephrol. 2017; 32(10): 1851–1859.Published online 2016 Oct 18. doi: 10.1007/s00467-016-3499-x
PMCID: PMC5579149 PMID: 27757584
Zaniew M, Mizerska-Wasiak M, Załuska-Leśniewska I, Adamczyk P, Kiliś-Pstrusińska K, Haliński A, Zawadzki J, Lipska-Ziętkiewicz BS, Pawlaczyk K,Sikora P, Ludwig M, Szczepańska M.Dent disease in Poland: what we have learned so far? Int Urol Nephrol. 2017;49(11):2005-2017. doi:10.1007/s11255-017-1676-x. Epub 2017 Aug 16. PMID: 28815356
Deng H, Zhang Y, Xiao H, Yao Y, Zhang H, Liu X, Su B, Guan N, Zhong X, WangS, Ding J, Wang F. Phenotypic spectrum and antialbuminuric response to angiotensin converting enzyme inhibitor and angiotensin receptor blocker therapy in pediatric Dent disease. Mol Genet Genomic Med. 2020Aug;8(8):e1306. doi: 10.1002/mgg3.1306. Epub 20203.PMID: 32495484
rarediseases.org/rare-diseases/dent-disease/
Ehlayel AM, Copelovitch L.Update on Dent Disease. Pediatr Clin North Am.2019;66(1):169-178. doi: 0.1016/j.pcl.2018.09.003.PMID: 30454742 Review.
Jin YY, Huang LM, Quan XF, Mao JH. Dent disease: classification, heterogeneity and diagnosis. World J Pediatr. 2021;17(1):52-57. doi:10.1007/s12519-020-00357-1. Epub 2020 Apr 4.PMID: 32248351
Liu J, Sadeh TT, Lippiat JD, Thakker RV, Black GC, Manson F. Small molecules restore the function of mutant CLC5 associated with Dent disease.J Cell MolMed. 202;25(2):1319-1322. doi: 10.1111/jcmm.16091.Epub 2020 Nov 16.PMID: 33200471