INNEHÅLLSFÖRTECKNING
GENETIK, ELLER ÄRFTLIGHET FÖR DENTS SJUKDOM
- Lågmolekylär proteinuri
- Hyperkalciuri
- Njurförkalkningar och njursten (nefrolithiasis)
- Hematuri
- Fanconis syndrom
- Kronisk njursvikt
- Andra symtom

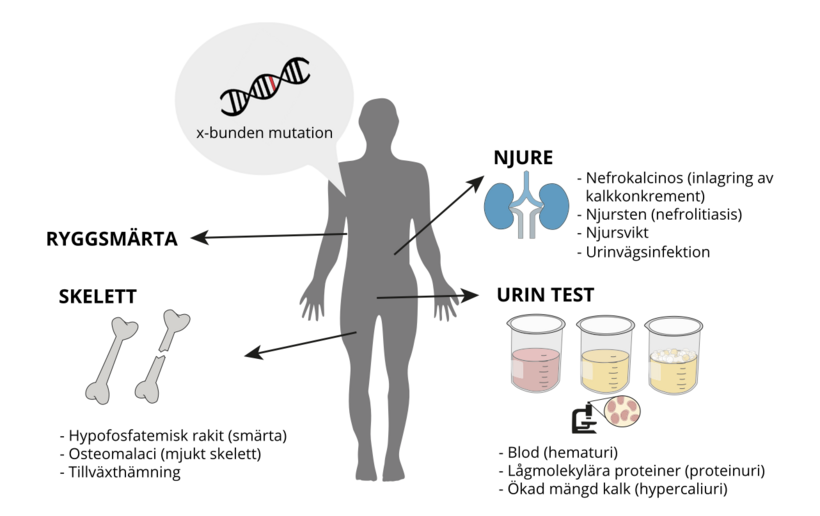
Dents sjukdom är en sällsynt medfödd njursjukdom som leder till utveckling av njursten och ofta även till njursvikt. Symtomen uppträder vanligtvis i barndomen, men de kan också gå obemärkta förbi fram till vuxen ålder. Sjukdomen kännetecknas främst av förekomsten av små proteinmolekyler i urinen i kombination med hyperkalciuri, dvs. ökad utsöndring av kalcium i urinen, bildning av kalciumavlagringar i njurarna (nefrokalcinos) och återkommande episoder av njursten (nefrolithiasis).
De progressiva njurproblemen leder ofta till njursvikt i vuxen ålder. Mindre vanligt är rakit, dvs. skelettförändringar till följd av kalcium-fosfatrubbningar, och tillväxtstörningar.
Sjukdomen drabbar i allmänhet endast män, eftersom den är kopplad till könskromosomen (se nedan). Förekomsten av sjukdomen kan vara underskattad, förmodligen på grund av att många patienter inte berättar om sina besvär och att sjukdomen är föga känd. Sjukdomen diagnostiseras ofta för sent då njurfunktionen redan är påverkad.
Dents sjukdom är en medfödd genetiskt betingad sjukdom. Den är monogen, vilket innebär att dess uppkomst beror på en mutation i en enda specifik gen (i detta fall antingen CLCN5- eller OCRL1-genen). Dents sjukdom tillhör den grupp av sjukdomar som kallas tubulopatier. Tubulopatier är sjukdomar där en defekt påverkar tubuli i njurarna (se nedan).
Man skiljer mellan proximala, distala och blandade tubulopatier, beroende på vilken del av tubuli som är drabbad. Vid Dents sjukdom påverkar funktionsstörningen främst de proximala tubuli. I denna del av tubuli återabsorberas många ämnen som har filtrerats i glomeruli (njurens filter) eftersom kroppen fortfarande behöver dem. Generna CLCN5 och OCRL1 kodar för specifika enzymer som är nödvändiga för återabsorptionen av lågmolekylära proteiner och mineraler som kalium, fosfat, kalcium och bikarbonat.
Visste du att?
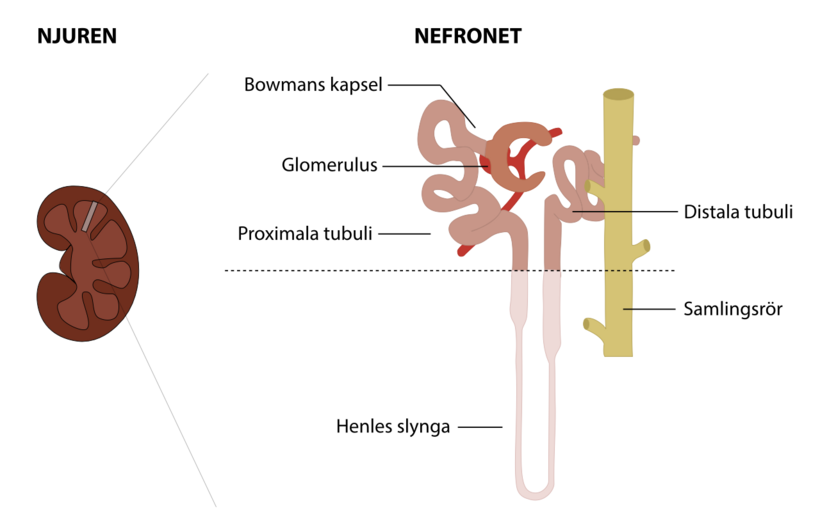
Njurarnas huvuduppgift är att avlägsna överflödigt vatten och avfallsprodukter från kroppen genom att bilda urin. Varje njure innehåller i genomsnitt cirka en miljon nefron, som var och en består av ett filter (glomerulus) och ett rör som kallas tubulus. I glomerulus bildas primärurin genom filtrering av cirkulerande blod. Filtret är normalt ogenomträngligt för blodkroppar och stora proteiner.
Tubuli behövs för att återvinna ämnen som har filtrerats i glomeruli men som är värdefulla för kroppen, t.ex. vatten, elektrolyter (som natrium, klorid, kalium, kalcium, magnesium, fosfor och många andra), glukos, aminosyror och proteiner. De reglerar också syra-bas balansen. Dessa processer är nödvändiga för att upprätthålla en stabil balans av kroppskemikalier. Hos en vuxen person filtrerar njurarna ca 150 liter vatten per dag, varav 99% återabsorberas i tubuli, vilket ger ca 1,5 liter slutlig urin. Detta är möjligt tack vare att njurtubuli har en total längd på 80 km!
Tubuli består av följande segment: proximala tubuli, Henles slynga, distala tubuli och samlingsröret. I proximala tubuli sker huvuddelen av reabsorptionen. Njurarna producerar också hormoner som påverkar funktionen hos andra organ. Till exempel det hormon som stimulerar produktionen av röda blodkroppar. Andra hormoner som produceras av njurarna hjälper till att reglera blodtrycket och kontrollera kalciummetabolismen.

Orsaken till sjukdomen är en mutation i en enda gen, CLCN5 eller OCRL1. Båda dessa gener finns på könskromosomen X, vilket innebär att nedärvningen av dessa mutationer och sjukdomen är kopplad till kön.
Hos en man innehåller varje kroppscell en X-kromosom och en Y-kromosom, hos en kvinna två X-kromosomer.
Dents sjukdom nedärvs recessivt kopplat till X-kromosomen. Detta innebär att sjukdomen vanligtvis endast drabbar män, eftersom kvinnor har två X-kromosomer, varav den ena kan kompensera för defekten hos den andra X-kromosomen.
Sjukdomen manifesterar sig endast om en person inte har minst en frisk X-kromosom.
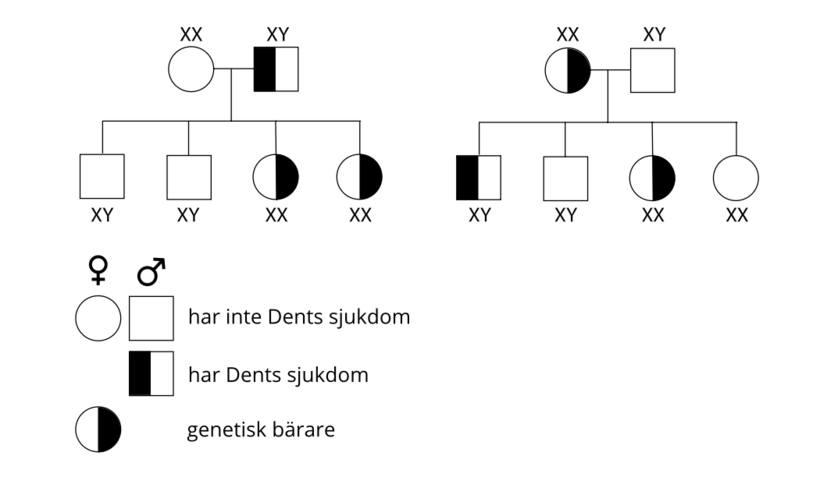
Om en mamma är bärare av CLCN5- eller OCRL1-mutationen är risken 50 procent att hon överför mutationen till sina barn av båda könen. De flickor som får en muterad X-kromosom från sin mor blir dock inte sjuka eftersom de får en frisk X-kromosom från sin far. Pojkar som får en muterad X-kromosom blir dock sjuka eftersom de ärver en Y-kromosom från sin far och inte har någon frisk X-kromosom.
En kvinna som har en muterad och en frisk X-kromosom kallas bärare av sjukdomen eftersom hon inte har några symtom men kan föra sjukdomen vidare till sina barn. Eftersom det bara behövs en enda aktiv X-kromosom är en av de två inaktiverad hos kvinnor. Detta är en slumpmässig händelse som inträffar i varje kroppscell separat. Kvinnor som är bärare av sjukdomen har därför en blandning av muterade och omuterade celler. I vissa fall kan kvinnor ha en fördelaktig X-inaktivering där den muterade X-kromosomen tystas i de flesta celler. Dessa kvinnor kanske inte utvecklar några eller endast mycket milda symtom på sjukdomen.
I andra fall kan kvinnor ha en negativ X-inaktivering, dvs att den friska X-kromosomen tystas ned i de flesta celler. Drabbade kvinnor kan därför utveckla varierande symtom på Dents sjukdom.
I extremt sällsynta fall uppstår mutationen slumpmässigt utan att någon av föräldrarna bär på mutationen. Detta kallas en “de novo-mutation” och innebär att mutationen uppstår i en könscell (ägg eller spermie) hos en av föräldrarna eller i själva det befruktade ägget under tidig embryogenes.
I denna situation uppträder förändringen för första gången hos just detta foster och övriga barn till dessa föräldrar kommer att vara opåverkade. Den drabbade individen kan dock komma att föra mutationen vidare till sina barn. Att fastställa mutationens ursprung (de novo eller ärftlig) kan vara viktigt i vissa situationer, exempelvis när föräldrar med ett sjukt barn planerar en ny graviditet eller om en mamma vill donera en njure åt en son som utvecklat njursvikt till följd av Dents sjukdom.
De flesta celler innehåller en komplett uppsättning gener. Gener är “livets recept” och fungerar som en uppsättning instruktioner som styr vår tillväxt och hur vår kropp fungerar. De är ansvariga för många av våra egenskaper, t.ex. ögonfärg eller kroppslängd. När en genmutation uppstår kan proteinprodukten vara defekt, ineffektiv eller saknas helt. Beroende på funktionen hos det specifika proteinet kan detta påverka ett eller flera organsystem. Generna består av ett kemiskt ämne som kallas DNA och finns inuti trådformiga strukturer som kallas kromosomer.
Varje människa har 46 kromosomer i de flesta celler. Dessa är 22 par autosomala kromosomer och 1 par könskromosomer, dvs. X och Y. Kromosomerna ärvs från föräldrarna, 23 från mamman och 23 från pappan, så varje person har 2 fullständiga uppsättningar av 23 kromosomer eller 23 “par”. Eftersom kromosomerna är uppbyggda av gener ärver alla 2 kopior av de flesta gener, en kopia från varje förälder. Situationen är något annorlunda när det gäller könskromosomer, där det vid manligt kön finns en X- och en Y-kromosom respektive två X-kromosomer vid kvinnligt kön.

Denna typ av nedärvning gäller gener som finns på autosomala kromosomer, dvs. som inte är relaterade till kön. Vid dominant nedärvning räcker det med en kopia av den defekta genen för att sjukdomssymtomen ska uppstå. Vid recessiv nedärvning förhindrar en korrekt version av genen att sjukdomen manifesteras, dvs. det krävs två defekta gener för att sjukdomen ska manifesteras.
Vi talar om X-kromosombunden nedärvning när de gener vars mutationer orsakar en viss sjukdomsenhet finns på X-kromosomen. Dessa sjukdomar skiljer sig från autosomala sjukdomar genom att de drabbar kvinnor och män på olika sätt. Detta beror på att båda könen har olika uppsättningar av könskromosomer. Medan kvinnor har två X-kromosomer, och därmed två versioner av varje gen på X-kromosomen, har män bara en enda X-kromosom, dvs. bara en version av en viss gen. En defekt gen på X-kromosomen leder därför till sjukdom hos en man, medan den andra X-kromosomen kan kompensera hos en kvinna (X-kromosombunden recessiv nedärvning).
Dents sjukdom typ 1, 2 och 3
Hittills har två typer av sjukdomen identifierats. Båda formerna är ärftliga i ett X-kromosombundet recessivt sätt, men mutationerna involverar två olika gener.
Orsaken till sjukdomen är en mutation i CLCN5-genen, som bestämmer funktionen hos kloridkanalen CLC-5. CLC-5 har en nyckelroll i försurningen i endosomerna i proximala tubulära celler, vilket är viktigt för återabsorptionen av proteiner med låg molekylvikt (LMW) från urinen. Detta är den vanligaste typen av sjukdomen och står för cirka 65 procent av alla fall av Dents sjukdom. Det finns många olika mutationer av denna gen och symtomen kan variera avsevärt även hos patienter som har samma mutation.
Typ 2 förekommer hos cirka 10-15% av patienterna med Dents sjukdom och orsakas av mutationer i OCRL1-genen. Denna typ av sjukdom kännetecknas av samma njursymtom som förekommer vid typ 1, men dessutom kan symtom som lindrig intellektuell funktionsnedsättning, ögonförändringar i form av lindrig katarakt (en grumling av den normalt klara linsen) och kortvuxenhet förekomma. OCRL-genen kodar för ett protein som är involverat i intracellulär transport i proximala tubuliceller, men också i många andra processer i kroppen. Vissa typer av mutationer i OCRL-genen orsakar Dents sjukdom typ 2, medan andra orsakar en mycket allvarligare sjukdom som kallas Lowes syndrom (se nedan).
Den sjukdom som de patienter har, som har drag av Dents sjukdom men där ingen av ovanstående mutationer kunnat påvisas, kallar man Dents sjukdom typ 3. Denna grupp utgör 25-35% av alla patienter med Dents sjukdom och det är troligt att andra, ännu oidentifierade, gener orsakar denna sjukdom.
Dents sjukdom beskrevs första gången av Charles Enrique Dent och M. Friedman 1964. De rapporterade om två obesläktade brittiska pojkar med rakit och tecken på renal tubulär dysfunktion i form av hyperkalciuri, hyperfosfaturi, proteinuri och aminoaciduri (aminoaciduri - se också termen i ordlistan - onormalt höga mängder aminosyror i urinen). Namnet på sjukdomen gavs 30 år senare, när njurläkaren Oliver Wrong mer utförligt beskrev sjukdomen och valde att namnge sjukdomen efter sin mentor. Sjukdomen drabbar nästan uteslutande män och symtom kan uppträda redan i tidig barndom.
Svårighetsgraden av Dents sjukdom kan variera kraftigt, även bland drabbade familjemedlemmar. Drabbade individer behöver inte ha alla de symtom som diskuteras nedan. I varje specifikt fall bör de förekommande avvikelserna, deras behandling samt den allmänna prognosen bedömas individuellt. Ibland diagnostiseras sjukdomen slumpmässigt när fortfarande inga symptom föreligger, men vanligare är att patienten diagnostiserats med avancerad kronisk njursjukdom av okänd etiologi i åldern 30-50 år. Kvinnor som bär på de någon av de mutationer som hos män orsakar Dents sjukdom kan ha mild småmolekylär proteinuri och hyperkalciuri men njursten och njursvikt är sällsynt.
Proteinuri med låg molekylvikt (LMW-proteinuri)
Vanligtvis är utsöndring av beta 2-mikroglobulin i urinen mer än 5-faldigt ökad!
Vid Dents sjukdom varierar symptom och fynd, men proteinuri förekommer alltid. Den typ av proteinuri som observeras vid Dents sjukdom kallas lågmolekylär proteinuri (LMW-proteinuri).
LMW-proteiner är små proteiner som filtreras i glomeruli men återabsorberas i proximala tubuli så att de vanligtvis inte kan detekteras i urinen hos friska personer. Mätning av LMW-proteiner kräver specifika tester och de kan missas vid rutinmässig testning.
Exempel på LMW-proteiner som används för diagnostik är beta-2-mikroglobulin, alfa-1-mikroglobulin och retinolbindande protein. Förekomst av ett LMW-protein i urinen indikerar nedsatt funktion i de proximala tubuli. Vid Dents sjukdom är urinkoncentrationen av beta-2-mikroglobulin minst fem gånger högre än den övre gränsen för normalvärdet. För kvantitativ bedömning av lågmolekylär proteinuri kan du utföra en 24-timmars urinsamling eller bedöma förhållandet mellan beta-2-mikroglobulin (som ett exempel på ett LMW-protein) och kreatinin i en slumpmässig urinportion. Eftersom förekomst av proteiner i urinen är ett vanligt symptom på störningar i glo- meruli snarare än tubuli, kan läkare ibland missta Dents sjukdom för en form av glomerulonefrit (inflammation i glomeruli) eller ett nefrotiskt syndrom (massivt läckage av proteiner genom det glomerulära filtret). En mer detaljerad analys av urinen visar dock att majoriteten av proteinerna i urinen hos patienter med Dents sjukdom utgörs av LMW-proteiner.
Fanconis syndrom och låga fosfatnivåer i serum
Nedsatt reabsorption av proteiner med låg molekylvikt i de proximala njurtubuli förekommer ofta tillsammans med nedsatt reabsorption av andra ämnen som fosfat, kalium, aminosyror eller bikarbonater. Beroende på antalet substanser som påverkas kallas detta ett ofullständigt eller fullständigt renalt Fanconis syndrom.
Komplett Fanconis syndrom (De Toni-Debré-Fanconis syndrom) innebär en defekt som påverkar alla funktioner i de proximala tubuli och orsakar förlust av aminosyror, glukos, fosfat, urinsyra, citrat, proteiner med låg molekylvikt, magnesium, kalium, kalcium, bikarbonat och vatten.
Med inkomplett Fanconis syndrom avses förlust av endast några av de ovan nämnda komponenterna. Hos de flesta patienter med Dents sjukdom påverkas inte alla funktioner i proximala tubuli. Pågående urinförluster leder till minskade blodkoncentrationer av de berörda ämnena: fosfatförluster leder till hypofosfatemi, kaliumförluster till hypokalemi och bikarbonatförluster till acidos. Förlusten av aminosyror har inga metabola konsekvenser. Eftersom fosfat krävs för benbildning kan hypofosfatemi orsaka rakit eller osteomalaci (se ordlistan), som (till skillnad från andra former av rakit) inte svarar på höga doser av vitamin D. Benskadorna förvärras av acidosen, som orsakar benresorption. I slutändan kan tillväxten försämras och patienterna kan utveckla bendeformiteter. Hypokalemi kan orsaka muskelsvaghet och försämra vattenreabsorptionen, vilket leder till ökad urinproduktion (polyuri) och törst (polydipsi), vilket kan leda till uttorkning.
Hyperkalciuri - ökad utsöndring av kalcium i urinen
Hyperkalciuri, liksom LMW-proteinuri, kan endast upptäckas genom laboratorietester. För ögat brukar urinen se normal ut, även om hyperkalciuri kan åtföljas av hematuri, dvs förekomst av blod i urinen. Orsaken till hyperkalciuri vid Dents sjukdom är ännu inte helt klarlagd. En potentiell mekanism är att parathormon, ett lågmolekylärt protein som påverkar kalciumutsöndringen, inte reabsorberas. Även förlust av ett protein som binder D-vitamin kan vara inblandad. En annan mekanism kan vara ökad kalciumfrisättning från skelettet på grund av metabol acidos, ett annat kännetecken för Dents sjukdom. För att bedöma utsöndringen av kalcium beräknas förhållandet mellan kalcium- och kreatininkoncentrationen i en portion av urinen.
Förkalkningar i njurarna (nefrokalcinos) eller njursten (nefrolithiasis)
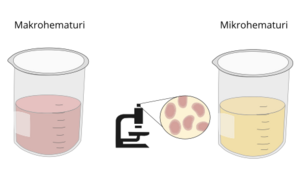
Ökad kalciumkoncentration i urinen leder till kristallisering som i sin tur leder till förkalkningar i njurvävnaden (nefrokalcinos) eller att njursten (nefrolithiasis) bildas. Njurförkalkningar och njursten kan visualiseras med ultraljud. Blod i urinen – ibland synlig endast med hjälp av urinmikroskopi (“mikrohematuri”) – är ibland är det första symtomet på njursten. Njursten kan också orsaka andra symtom som smärtsam blåstömning (dysuri), en önskan att kissa ofta, buksmärtor (renal kolik), obstruktion av urinflödet och/eller mupprepade urinvägsinfektioner.
Kronisk njursjukdom och njursvikt
Sjukdomsförloppet kan leda till kronisk njursjukdom (CKD) med en gradvis försämring av njurfunktionen. Exempel på symtom förenliga med avancerad kronisk njursjukdom är aptitlöshet, oavsiktlig viktminskning, trötthet och anemi. I vissa fall, ibland så tidigt som i 30-50-årsåldern, kan de drabbade personerna utveckla njursvikt och behöva dialys eller njurtransplantation.
Andra symtom
Vissa personer med Dents sjukdom kan också utveckla bensjukdom som osteomalaci och hypofosfatemisk rakit, ett tillstånd som orsakas av försämrad fosfattransport och förändrad D-vitaminmetabolism i njurarna.
Hos barn med Dents sjukdom kan tillväxttakten vara långsammare än normalt vilket ofta resulterar i måttlig kortvuxenhet. Barnen kan också uppleva smärta i benen och ha svårt att gå. På grund av skelettförändringar kan både barn och vuxna ha en ökad risk för frakturer.
Vissa personer med Dents sjukdom har brist på vitamin A, vilket kan leda till försämrat mörkerseende och torra ögon (xeroftalmi). Vitamin A-brist orsakas i detta fall av förlust i urinen av ett lågmolekylärt protein som binder retinol. Symptomen kan behandlas med tillskott av vitamin A.
Vissa personer med Dents sjukdom typ 2 kan ha andra ytterligare symtom, t.ex. lindrig intellektuell funktionsnedsättning, minskad muskeltonus och försenad motorisk utveckling, samt grumling av ögonlinserna (katarakt), vilket vanligtvis inte försämrar synen.

Proteinuri med låg molekylvikt i kombination med hyperkalciuri eller nefrokalcinos eller njursten hos en pojke/man kan vara de enda symtomen på sjukdomen och bör leda till ytterligare diagnostik.
Den kliniska diagnostiken av Dents sjukdom baseras på identifiering av karakteristiska symtom, en detaljerad sjukdoms- och familjehistoria, en grundlig klinisk bedömning och olika specialtester.
På grund av sjukdomens varierande symtom bör Dents sjukdom också övervägas hos män som har:
- Symtom på glomerulär sjukdom (steroidresistent nefrotiskt syndrom)
- Dysfunktion i tubuli/Fanconis syndrom
- Idiopatisk nefrolithiasis
- Ospecificerad kronisk njursjukdom
Genetisk testning kan identifiera de två mutationer som är kända att orsaka Dents sjukdom, men är inte alltid nödvändig om en klinisk diagnos kan ställas (t.ex. lågmolekylär proteinuri och hyperkalciuri hos män). Vid nefrokalcinos och kronisk njursjukdom av oklar genes kan genetisk testning behövas för att skilja mellan Dents sjukdom och andra genetiska orsaker. Man bör komma ihåg att hos ungefär en tredjedel av männen med den typiska bilden av Dents sjukdom kan ingen mutation påvisas i någon av de två Dent-generna.
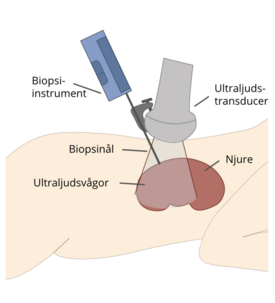
Njurbiopsi (ett litet prov av njurvävnaden som tas för mikroskopisk undersökning) utförs ofta på patienter med oförklarlig njursjukdom och/eller betydande proteinuri och hematuri. Av denna anledning kommer vissa patienter med Dents sjukdom ha genomgått en njurbiopsi innan man får sin diagnos. Biopsiresultaten är dock ospecifika och visar glomerulär skleros (FSGS) (se ordlistan), interstitiell fibros (ärrbildning) och nefrokalcinos.
Njurbiopsi är inte nödvändigt för att diagnostisera Dents sjukdom och kan till och med vara missvisande.
Kan Dents sjukdom förväxlas med någon annan sjukdom?
Det finns flera sällsynta genetiska sjukdomar som, liksom Dents sjukdom, kännetecknas av en benägenhet att bilda njursten. Exempel på sådana sjukdomar är primär hyperoxaluri, familjär hyperkalciuri med hypomagnesemi och nefrokalcinos (Michelis-Castrillos syndrom), brist på adeninfosforibosyltransferas (APRT) och cystinuri.
Primär hyperoxaluri (PH) | är en grupp sällsynta genetiska ämnesomsättningssjukdomar som kännetecknas av ackumulering av en substans som kallas oxalat i njurarna och andra organsystem i kroppen. Drabbade individer saknar ett enzym som normalt förhindrar ackumulering av oxalat. |
Familjär hyperkalciuri med hypomagnesemi och nefrokalcinos (Michelis-Castrillos syndrom, FHHNC) | är en sällsynt genetisk sjukdom som nedärvs autosomalt recessivt och kännetecknas av förlust av magnesium och kalcium i urinen. Sjukdomen kännetecknas av hypomagnesemi, hyperkalciuri och nefrokalcinos och patienterna uppvisar kliniskt polyuri, polydipsi och vitamin D-resistent rakit. FHHNC beror på mutationer i CLDN16- eller CLDN19-generna. CLDN16 och CLDN19 kodar för tight-junction-proteinerna claudin-16 respektive claudin-19, som uttrycks i den tjocka uppstigande delen av Henles slynga och bildar ett viktigt komplex för den paracellulära reabsorptionen av magnesium och kalcium. Dessa patienter har inte hypokalemi eller saltförlust. Patienter med mutationer i CLDN19 uppvisar också allvarliga ögonförändringar som närsynthet, nystagmus och kolobom. |
Brist på adeninfosforibosyltransferas (APRT) | kännetecknas av en förhöjd produktion och renal utsöndring av 2,8-dihydroxyadenin (DHA), vilket leder till bildning av njursten och kristallinducerad njurskada (dvs. nefropati med DHA-kristaller) som orsakar episoder av akut njursvikt och progressiv kronisk njursjukdom. |
Cystinuri | är en autosomal recessiv ärftlig sjukdom som kännetecknas av höga koncentrationer av aminosyran cystin i urinen, vilket leder till bildandet av cystinstenar i njurar och urinvägar. |
Fanconi Syndrom | En rad ärftliga sjukdomar kännetecknas av Fanconis syndrom och bör därför också beaktas vid differentieringen av Dents sjukdom. Som exempel kan nämnas genetiska sjukdomar som cystinos, ärftlig fruktosintolerans, galaktosemi, tyrosinemi, Wilsons sjukdom och olika glykogenoser. Renalt Fanconis syndrom kan också förvärvas under livet som en biverkning av vissa läkemedel (t.ex. valproat, deferasirox, cisplastin, ifosfamid) eller vara sekundärt till vissa njursjukdomar, cancerformer som multipelt myelom, Sjögrens syndrom eller hyperparatyreoidism. Hos barn kan exponering för tungmetaller också orsaka Fanconis syndrom. |
Lowes syndrome | Lowe’s syndrome, is a rare genetic disorder characterized by vision problems, including clouding of the eye lenses (cataracts) that are present at birth, kidney problems that usually develop in the first year of life, and brain abnormalities associated with intellectual disability. The specific symptoms and severity of the disorder can vary greatly from person to person. Lowe syndrome is inherited as an X-linked trait. Lowe syndrome is caused by mutations in the same gene (OCRL1) that causes Dent’s disease type 2. The disorder is fully expressed only in men. är en sällsynt genetisk sjukdom som kännetecknas av synproblem, inklusive grumling av ögonlinserna (katarakt) som finns vid födseln, njurproblem som vanligtvis utvecklas under det första levnadsåret och hjärnabnormaliteter som är förknippade med intellektuell funktionsnedsättning. De specifika symtomen och sjukdomens svårighetsgrad kan variera kraftigt från person till person. Lowes syndrom nedärvs som en X-kromosombunden egenskap. Lowes syndrom orsakas av mutationer i samma gen (OCRL1) som orsakar Dents sjukdom typ 2. Sjukdomen är fullt uttalad endast hos män. |
Om proteinuri är det första symtomet kan Dents sjukdom lätt förväxlas med sjukdomar som påverkar glomerulus (glomerulonefrit och nefrotiskt syndrom). Dessa sjukdomar behandlas ofta med kortikosteroider, vilket inte hjälper eftersom Dents sjukdom inte är en inflammation i njurarna utan en genetisk sjukdom. Det faktum att steroider inte hjälper kan leda till att läkaren klassificerar Dents sjukdom som ett “steroidresistent nefrotiskt syndrom” innan rätt diagnos har ställts. Det kan vara särskilt vilseledande när en njurbiopsi görs och visar fokal segmentell glomeruloskleros (FSGS).

Vem kommer att delta i min/mitt barns behandling?
Behandling av patienter med Dents sjukdom bör utföras av, eller under handledning av, njurmedicinsk expertis och syftar vanligtvis till att lindra symtom med hjälp av läkemedel som hämmar bildandet av njursten samt kosttillskott som korrigerar elektrolyt- och metabola störningar.
Det behövs ofta ett nära samarbete mellan olika specialister: barnläkare, njurläkare, urologer, dietister och annan hälso- och sjukvårdspersonal.
Behandlingen består av:
- Minimera kalciuminlagringar i njurar och urinvägar, dvs förhindra bildandet av nefrokalcinos och njursten (profylaktisk behandling)
- Avlägsnande av stenar från urinvägarna (symtomatisk behandling)
- Behandling av sjukdomens effekter som exempelvis elektrolytrubbningar och progressiv kronisk njursvikt
Tillräcklig vätskeintag >3 liter/m² kroppsyta
Tillräckligt vätskeintag, i detta fall mer än genomsnittligt, är en viktig åtgärd som minskar kalciuminlagringar i njurarna. Sannolikt minskar effekten av andra förebyggande behandlingar om man inte får i sig tillräckligt med vätska. Det dagliga vätskebehovet skattas utifrån kroppsytan (body surface area, BSA) som beräknas med hjälp av kalkylatorer som finns tillgängliga online eller med hjälp av formeln:
BSA = √(Kroppsvikt (kg) x längd (cm)/ 3600)
Glöm inte att behovet av vätska ökar i situationer med diarré, feber, kräkningar eller om du befinner dig i ett varmt klimat. Vid ökad vätskeförlust eller minskat vätskeintag bör man skyndsamt kontakta sjukvården för rådgivning i syfte att förhindra onödig försämring av sjukdomen. Om man inte får i sig tillräckligt mycket vätska via munnen kan det i vissa fall bli nödvändigt med intravenös vätsketillförsel. Patienter, vårdgivare och lärare behöver vara medvetna om detta.
Vänligen observera!:
Ha alltid med dig ett dokument med information om din/ditt barns sjukdom
- Det dagliga vätskeintaget bör överstiga 2,5-3 liter/m² kroppsyta
- Se till att du alltid har gott om vätska till ditt förfogande
- Drick även om du inte är törstig - med jämna mellanrum under hela dagen
- Planera in täta toalettbesök
Du kan ställa in påminnelser på din mobiltelefon för att komma ihåg att dricka vatten, du kan också använda speciella appar för att registrera hur mycket vatten du dricker.
Diet
Begränsat intag av bordssalt minskar mängden kalcium som utsöndras i urinen och minskar därmed risken för njurstensbildning.
Behandling med läkemedel
Tillskott av kalium och fosfat
Eftersom Dents sjukdom kännetecknas av tubulär förlust av viktiga ämnen som exempelvis kalium måste dessa tillföras för att hålla blodkoncentrationerna inom det normala intervallet. Hos patienter med låga fosfatnivåer (hypofosfatemi) och rakit förskrivs fosfattillskott. Fosfattillskott kan minska hyperkalciuri, även i frånvaro av hypofosfatemi. Läkemedel som minskar kalciumkristalliseringen (i kombination med vätskeintag) kan ytterligare minska risken för stenbildning.
De bästa resultaten uppnås när läkemedlen tas med jämna mellanrum under dagen.
Kaliumcitrat bildar lösliga komplex med kalcium i urinen, vilket minskar tillgången på kalcium för kristallbildning. Citrat omvandlas i levern till bikarbonat och leder till mer alkaliskt blod och urin (högre pH-värde i blod och urin). Under dessa förhållanden återabsorberas mindre citrat i njurtubuli och mer utsöndras i urinen. Citrat binder kalcium och minskar tillgängligheten av kalcium för stenbildning. Doseringen av alkaliskt citrat justeras individuellt baserat på resultatet av urinprovets pH, vilket kan testas med hjälp av pH-indikatorpapper. PH-värdet bör ligga mellan 6,2 och 7,4. Kronisk behandling med citrat kan fördröja utvecklingen av njursjukdom och förhindra bildandet av stenar.
Tiaziddiuretika förskrivs ofta vid Dents sjukdom för att minska hyperkalciuri. Dess användning begränsas dock av biverkningar som hypovolemi (minskad kroppsvätska, vattenförlust) och hypokalemi (sänkt kaliumnivå i serum). Därför måste denna behandling övervakas noga av den behandlande njurläkaren och extra försiktighet rekommenderas vid minskat vätskeintag eller ökade vätskeförluster.
ACE-hämmare förskrivs av vissa njurläkare i ett försök att minska proteinuri. Dessa läkemedel sänker filtreringstrycket i glomeruli och därmed mängden protein som passerar genom de glomerulära filtren. Denna behandling är kontroversiell eftersom problemet vid Dents sjukdom är minskat upptag av proteiner i tubulus och inte ökad filtration i glomeruli. Hittills har inga studier utförts för att testa om ACE-hämmare är bra vid Dents sjukdom.
är en minimalt invasiv metod för att avlägsna stenar från urinvägarna med hjälp av laser. Under anestesi förs ett endoskop in i urinblåsan via urinröret och vidare upp i urinledaren till stenen som fragmenteras med lasern. Om fragmenten som bildas är små får de passera spontant, annars tas de ut med hjälp av endoskopet.
Även idag diagnostiseras Dents sjukdom ofta först när njurfunktionen redan är kraftigt nedsatt. I dessa fall krävs behandling av kronisk njursvikt och dess komplikationer.
Bibehålla njurfunktionen så länge som möjligt
För att bibehålla njurfunktionen så länge som möjligt är det viktigt att vara särskilt uppmärksam på faktorer som kan vara skadliga för njurfunktionen. Av denna anledning rekommenderas följande:
Undvik läkemedel som kan skada njurarna, t.ex. icke-steroida antiinflammatoriska läkemedel (”NSAID”) - såsom Ibuprofen, Naproxen etc. som finns receptfria på många apotek. Det finns ett antal andra läkemedel som bör undvikas - din behandlande läkare kan berätta för dig om detta.
Mycket begränsad användning av, alternativt undvik helt, kontrastmedel vid radiologiska undersökningar som datortomografi (CT-scan).
Oavsett sammanhang ska du alltid berätta för din läkare att du har Dents sjukdom, så att han/hon kan justera behandlingen eller planera in extra kontroller vid behov.
Njurtransplantation
Vid njursvikt i slutstadiet, när njurarna inte kan utsöndra tillräckligt med slaggprodukter för att hålla kroppen vid liv, påbörjas dialys eller utförs en njurtransplantation.
Dialys
Dialys är ett förfarande där njurarnas grundläggande funktioner, dvs. avlägsnande av vatten och slaggprodukter, utförs av en maskin. Det finns två typer av dialys: hemodialys och peritonealdialys.
Vid hemodialys pumpas blodet genom ett filter där det renas och överflödig vätska avlägsnas. Denna metod utförs vanligtvis på sjukhus, t.ex. 4 gånger i veckan under flera timmar.
Peritonealdialys innebär upprepad tillförsel och borttagning av dialysvätska in i/ ut ur bukhålan med hjälp av en kateter. Denna metod kan utföras hemma med hjälp av en speciell maskin, vanligtvis under natten medan patienten sover.
Hittills har ingen botande behandling för Dents sjukdom utvecklats. På grund av de många olika symtom som förekommer finns det inte heller någon standardbehandling. Eftersom Dents sjukdom är så sällsynt saknas kliniska prövningar på en stor grupp patienter för att bevisa effekten av läkemedel som citrat eller tiazider. Försök med djurmodeller av Dents sjukdom har dock gett viktig kunskap. Forskning om genterapi för behandling av Dents sjukdom pågår. Experimentella data visar att den defekta funktionen hos det CLC5-protein som är muterat vid Dents typ 1 sjukdom eventuellt kan återfås genom behandling med så kallade små molekyler.
E. Emergencies
Patienter med Dents sjukdom löper hög risk för uttorkning och akuta tillstånd med feber, diarré/kräkningar kan kräva behandling och vätsketerapi på sjukhus. Det är också viktigt att noggrant övervaka elektrolyter. Om patienterna har njursten kan detta leda till obstruktion av urinvägarna, vilket kräver akuta urologiska ingrepp för att förhindra försämring av njurfunktionen.
Informera kirurgen / anestesiologen om diagnosen “Dents sjukdom”. Preoperativ bedömning av njurfunktion och elektrolyter (i synnerhet kalium) bör utföras på alla patienter med Dents sjukdom. Kontakta om möjligt den (pediatriska) njurläkare som vårdar dig/ditt barn före operation eller andra medicinska ingrepp för att diskutera särskilda krav/behov eller restriktioner för dig/ditt barn.
Det oförutsägbara förloppet och risken för en plötslig försämring av njurfunktionen utgör en stor psykologisk börda - både för de drabbade patienterna och deras familjer. De flesta patienter, inklusive deras föräldrar, behöver psykologiskt stöd.

Vad blir de långsiktiga resultaten av behandlingen?
Enligt tidigare observationer av patienter med Dents sjukdom slutar njurarna att fungera mellan 30 och 50 års ålder hos 30-80% av männen med Dents sjukdom. På grund av att sjukdomen är så sällsynt saknas det uppgifter om hur en tidig diagnos påverkar den fortsatta prognosen. Det verkar dock som om tidiga insatser minskar utvecklingen av nefrokalcinos och stenbildning, vilket kan bromsa utvecklingen av njursvikt.

Hur och var kan jag få mer hjälp?
I många länder finns det stödgrupper för patienter med Dents sjukdom. Dents sjukdom är en sällsynt sjukdom som inte är helt känd. Utbyte av information och erfarenheter kan vara till stor hjälp för patienter och deras familjer. Patientstödgrupper och stiftelser anordnar möten, föreläsningar och semesterläger.
Besök gärna patientgruppen för Dents sjukdom här.

| ACE-hämmare | en klass av läkemedel som minskar filtreringstrycket i njurarna och är grundpelaren i behandlingen av glomerulära sjukdomar |
| Aminoaciduri | onormalt höga halter av aminosyror i urinen |
| Kreatinin | en slaggprodukt som cirkulerar i blodet, filtreras av njurarna och. utsöndras i urinen. Kreatinin är inte skadligt utan används som en indikator på njurarnas funktion: ju högre blodkoncentration av kreatinin, desto sämre njurfunktion |
Kronisk njursjukdom (CKD) | progressiv och irreversibel njurskada som kan leda till njursvikt inom månader eller år. Eftersom njurarna inte kan återbildas finns det ingen behandling som kan häva kronisk njursjukdom, men det finns behandlingar som bromsar sjukdomsförloppet om de sätts in i tid |
| Dialys | metod för att avlägsna slaggprodukter och överskottsvätska från blodet. Det finns två huvudtyper av dialys: hemodialys och peritonealdialys. Vid hemodialys pumpas blodet genom ett filter med hjälp av en maskin. Peritonealdialys innebär upprepad tillförsel och borttagning av dialysvätska i/ur buken, vilket också renar blodet |
Njursjukdom i slutstadiet (ESKD) | den allvarligaste formen av njursjukdom när njurarna har slutat fungera (kan dock fortfarande producera urin som är av mycket dålig kvalitet). Detta innebär att njurersättningsterapi (dialys eller njurtransplantation) behövs |
| Fanconis syndrom | en uppsättning symtom som orsakas av en defekt i den första delen av nefronet (proximala tubuli), vilket orsakar en störning i resorptionen av aminosyror (aminoaciduri), glukos, fosfat, urinsyra, citrat, små proteiner, magnesium, kalium, kalcium, bikarbonat och vatten |
Fokal segmentell glomeruloskleros (FSGS) | ett tillstånd där ärrvävnad utvecklas i njurarnas filter (glomeruli) som kan leda till njursvikt. FSGS visar sig vanligtvis genom stora mängder protein i urinen |
| Gen | den genetiska enhet som innehåller instruktionen (“receptet”) för hur varje protein ska produceras i kroppen |
Glomeruli | de små filtren i njuren i början av varje nefron. Varje njure innehåller mellan 250 000 och 1 miljon glomeruli |
Glomerulär filtrationshastighet (GFR) | beskriver den hastighet med vilken njurarna filtrerar bort avfallsprodukter från blodet. GFR är normalt högre än 90 ml/min/1,73 m2, ett lägre värde indikerar nedsatt njurfunktion. Ett värde under 30 ml/min/1,73 m2 motsvarar svår njursvikt, vid ca 10 ml/min/1,73 m2 krävs njurersättningsterapi |
Njurtransplantation | en operation där en frisk njure sätts in i en person vars njure har slutat fungera (njursjukdom i slutstadiet) |
| Lowes syndrome | okulo-cerebro-renalt syndrom, orsakat av mutationer i samma gen (OCRL1) som orsakar Dents sjukdom typ 2. Sjukdomen ärvs också X-kromosombundet recessivt och drabbar endast män |
| Makrohematuri | synligt blod i urinen |
| Mikrohematuri | förekomst av en liten mängd röda blodkroppar (erytrocyter) i urinen. Färgen på urinen är normal, erytrocyterna är endast synliga vid mikroskopisk undersökning |
Nefrokalcinos | förekomsten av många punktformiga förkalkningar i njurvävnaden som återspeglar avlagring av kalciumkristaller. Dessa förändringar är lätt synliga med ultraljud. Nefrokalcinos kan predisponera för utveckling av njursten |
| Nefron | är den grundläggande funktionella och strukturella enheten i njuren, den består av två delar: njurfiltret (glomerulus) och tubulus där reabsorptionen äger rum |
Nefrotiskt syndrom | ett tillstånd där njurfiltren läcker stora mängder proteiner, vilket leder till låg proteinhalt i blodet och vätskeansamlingar i kroppen, ofta med synligt svullna ögon och ben som följd |
| Osteomalaci | benuppmjukning orsakad av störd benmetabolism, främst på grund av otillräcklig tillförsel av fosfat, kalcium och vitamin D. Kan också orsakas av ökad kalciumfrisättning från benen |
| Polydipsi | överdrivet drickande. Detta kan vara ett symptom på ett antal sjukdomar som orsakar vattenförluster och resulterar i överdriven törst |
| Polyuri | onormalt stor urinproduktion (t.ex. mer än 3 liter per dygn hos vuxna) |
Njurtubuli | del av nefronet där den primära urinen från glomerulus modifieras genom resorption och utsöndring av molekyler. I tubuli modifieras ca 150 liter primärurin vilket resulterar i ca 1,5 liter urin som utsöndras i urinblåsan. Tubuli består av flera sektioner: proximala tubuli, Henles slynga, distala tubuli och samlingsröret |
Rakit | skelettförändringar som orsakas av störningar i kalcium-fosfatmetabolismen |
| Tubulopatier | sällsynta njursjukdomar där njurens tubulära funktion är nedsatt medan glomeruli fungerar normalt |
| Urolithiasis (nefrolithiasis) | bildning av stenar i urinvägarna (i njuren) |
Gianesello L, Del Prete D, Anglani F, Calò LA.Genetics and phenotypic heterogeneity of Dent disease: the dark side of the moon.Hum Genet.2021;140(3):401-421. doi: 10.1007/s00439-020-02219-2. Epub 2020 Aug 29. PMID: 32860533
Anglani F, Gianesello L, Beara-Lasic L, Lieske J. Dent disease: a window into calcium and phosphate transport. J Cell Mol Med. 2019;23:7132–7142. doi:10.1111/jcmm.14590.-DOI-PMC–PubMed
van Berkel Y, Ludwig M, van Wijk J, Bökenkamp A. Proteinuria in Dent disease: a review of the literature. Pediatr Nephrol. 2017; 32(10): 1851–1859.Published online 2016 Oct 18. doi: 10.1007/s00467-016-3499-x
PMCID: PMC5579149 PMID: 27757584
Zaniew M, Mizerska-Wasiak M, Załuska-Leśniewska I, Adamczyk P, Kiliś-Pstrusińska K, Haliński A, Zawadzki J, Lipska-Ziętkiewicz BS, Pawlaczyk K,Sikora P, Ludwig M, Szczepańska M.Dent disease in Poland: what we have learned so far? Int Urol Nephrol. 2017;49(11):2005-2017. doi:10.1007/s11255-017-1676-x. Epub 2017 Aug 16. PMID: 28815356
Deng H, Zhang Y, Xiao H, Yao Y, Zhang H, Liu X, Su B, Guan N, Zhong X, WangS, Ding J, Wang F. Phenotypic spectrum and antialbuminuric response to angiotensin converting enzyme inhibitor and angiotensin receptor blocker therapy in pediatric Dent disease. Mol Genet Genomic Med. 2020Aug;8(8):e1306. doi: 10.1002/mgg3.1306. Epub 20203.PMID: 32495484
rarediseases.org/rare-diseases/dent-disease/
Ehlayel AM, Copelovitch L.Update on Dent Disease. Pediatr Clin North Am.2019;66(1):169-178. doi: 0.1016/j.pcl.2018.09.003.PMID: 30454742 Review.
Jin YY, Huang LM, Quan XF, Mao JH. Dent disease: classification, heterogeneity and diagnosis. World J Pediatr. 2021;17(1):52-57. doi:10.1007/s12519-020-00357-1. Epub 2020 Apr 4.PMID: 32248351
Liu J, Sadeh TT, Lippiat JD, Thakker RV, Black GC, Manson F. Small molecules restore the function of mutant CLC5 associated with Dent disease.J Cell MolMed. 202;25(2):1319-1322. doi: 10.1111/jcmm.16091.Epub 2020 Nov 16.PMID: 33200471